
微软研究院近期推出了一款名为 skala 的深度学习交换-关联(xc)泛函,致力于为 kohn–sham 密度泛函理论(dft)提供一种高效且高精度的计算方法。

Skala 通过建模非局部电子效应,在保持与 meta-GGA 泛函相近计算成本的同时,实现了接近混合泛函的精度水平。在 W4-17 分子数据集上的原子化能测试中,其平均绝对误差(MAE)仅为 1.06 kcal/mol,而在单参考体系子集中更是低至 0.85 kcal/mol;在更广泛的 GMTKN55 基准测试中,加权均方根绝对误差(WTMAD-2)为 3.89 kcal/mol,显示出其精度可与当前最先进的混合泛函相媲美。
该泛函的设计聚焦于主族元素的高精度热化学性质预测,而非追求覆盖所有材料和体系的通用性。Skala 本身并未内嵌色散作用的学习模块,初版仍依赖固定的 D3(BJ) 色散校正方案。因此,它特别适用于需要半局部泛函计算开销但又要求混合泛函级别精度的分子化学任务,例如高通量反应能(ΔE)计算、反应势垒估计、构象与自由基稳定性排序,以及分子几何结构和偶极矩的预测等场景。
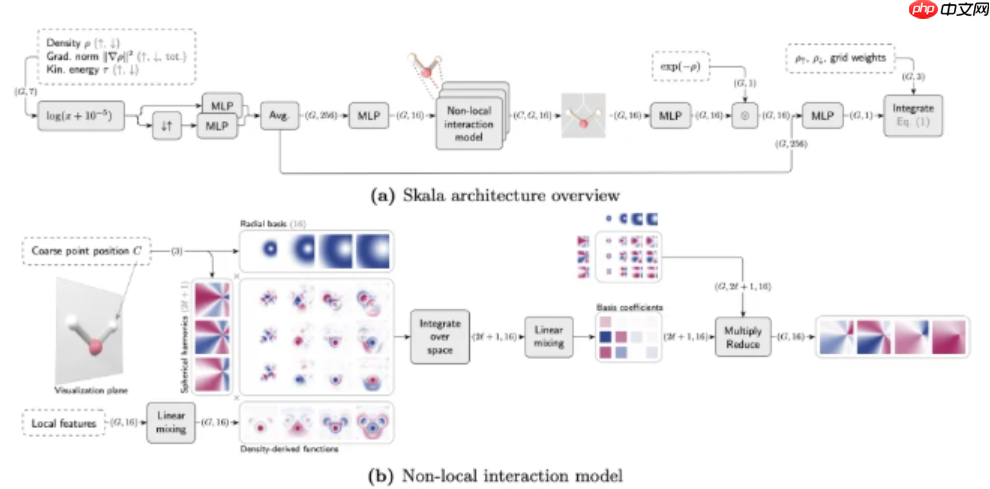
Skala 的模型架构与训练策略采用两阶段流程:第一阶段基于 B3LYP 计算得到的电子密度进行预训练,利用高精度波函数方法获得的 XC 能量作为目标标签;第二阶段则进入自洽场(SCF)内循环的微调过程,使用 Skala 自身生成的密度进行迭代优化,且无需反向传播支持 SCF 收敛。整个模型的训练依托一个大规模、高质量的数据集,包含约 80,000 个高精度总原子化能数据(MSR-ACC/TAE),确保了泛函的泛化能力与准确性。
为了保障实际应用中的效率,Skala 的算法复杂度控制在 O(N³),并针对 GPU 加速进行了专门优化。目前,该项目的开源代码和相关工具已发布于 Azure AI Foundry 实验室及 GitHub 平台,用户可在 PySCF、ASE 和 GauXC 等主流量子化学框架中直接调用,支持高效的批量 SCF 计算任务。

每个人都需要一台速度更快、更稳定的 PC。随着时间的推移,垃圾文件、旧注册表数据和不必要的后台进程会占用资源并降低性能。幸运的是,许多工具可以让 Windows 保持平稳运行。

 广告
广告Copyright 2014-2025 https://www.php.cn/ All Rights Reserved | php.cn | 湘ICP备2023035733号








 399
399
























